
How to Select the Right DAM Vendor for Your Technology Company
In life sciences, the path from clinical trial completion to regulatory approval is the final critical hurdle before patients can access new therapies. Yet this stretch, where regulatory, legal, quality, and medical affairs teams must collaborate to prepare submission documents, has become one of the industry’s most persistent bottlenecks.
The numbers tell the story. According to McKinsey’s research on regulatory submissions in pharmaceuticals, many organizations face months-long cycles to move from final clinical data to FDA submission-ready dossiers. A separate analysis of pharma review processes shows that promotional content review cycles commonly stretch to 50-60 days per item at mid-to-large pharmaceutical companies. These delays are driven not always by the review itself, but by fragmented submissions, missing documentation, and manual handoffs between teams. Gartner’s Market Guide for Life Science Regulatory Information Management Solutions highlights that legacy business processes and poor integration between systems remain the primary barriers to efficiency.
The industry is responding. McKinsey’s collaboration with Merck demonstrated that AI-powered platforms can reduce clinical study report (CSR) writing time from 180 hours to 80 hours while cutting errors by 50 percent. Beyond single tools, leading organizations are redesigning entire approval ecosystems, such as centralizing document management, automating metadata, and embedding compliance checks into every workflow stage. The result: some pharma companies have compressed regulatory timelines by 50 to 65 percent.
This transformation moves beyond “faster approvals.” It reframes how life sciences organizations think about legal and regulatory functions, shifting from gatekeepers that slow progress to compliance engines that accelerate safe, auditable submissions. As Aprimo explains in its guide to AI governance for enterprise content operations, effective compliance depends on embedding governance into workflows rather than treating it as a separate constraint.
TL;DR
- Regulatory submission and review cycles represent a critical bottleneck in life sciences, with traditional processes stretching 50-60 days or longer due to fragmented documents, incomplete submissions, and manual handoffs.
- Leading organizations have achieved 50-65 percent timeline compression through AI-enabled automation, workflow redesign, and centralized document management integrated with regulatory and quality systems.
- Modern solutions embed compliance and governance directly into content workflows—automating metadata, flagging missing data, triggering reviews, and maintaining audit trails, rather than treating approval as a separate review step.
- Centralized governance platforms enable cross-functional alignment between regulatory affairs, legal, medical affairs, quality, and clinical operations while maintaining 21 CFR Part 11, EU Annex 11, and other regulatory compliance requirements.
- Organizations that transform approval cycles from legal bottlenecks into compliance engines gain competitive advantages through faster time-to-market, fewer first-cycle deficiencies, and stronger regulatory relationships with health authorities.
The Regulatory Submission Bottleneck: Why Traditional Processes Fail
Regulatory submissions in life sciences have historically been complex, but today’s pressures are unprecedented. According to McKinsey’s analysis of pharmaceutical product development, developing a new molecule costs more than $4 billion (including failed candidates), and only 13 percent of candidates entering Phase I trials reach approval. This investment, measured in billions and years, makes every day lost to administrative delays painfully expensive.
The conventional submission process relies on a complex sequence of manual steps:
Document Assembly: Clinical, CMC (chemistry, manufacturing, and controls), safety, and efficacy data must be compiled, formatted, and cross-referenced according to FDA eCTD or EMA CTD standards. A typical regulatory dossier contains thousands of pages across multiple modules, each requiring precise organization and linking.
Multi-Team Review: Regulatory professionals, medical writers, quality experts, legal reviewers, and CMC specialists each evaluate portions of the submission. These reviews happen sequentially or, at best, with significant coordination overhead. Missing information at any stage triggers rework.
Compliance Verification: Every submission must satisfy dozens of regulatory requirements such as formatting rules, data integrity checks, completeness validations, often verified manually or through ad hoc processes.
Version Control Challenges: As documents cycle through review and revision, maintaining version control, tracking approvals, and ensuring the correct final versions are submitted, becomes error-prone.
The result: traditional timelines stretch 90-180 days from clinical database lock to submission-ready dossier. As one analysis of pharma regulatory challenges notes, regulatory submissions have become a structural bottleneck between innovation and patient access.

How Leading Organizations Are Redesigning Approval Workflows
Organizations at the forefront of regulatory efficiency are applying three core principles: centralization, automation, and integration.
Principle 1: Centralize Document Management with Compliance Built In
Rather than managing submissions across email, shared drives, and disconnected systems, leading organizations use centralized platforms that embed regulatory compliance into the system itself. According to Gartner’s guidance on regulatory information management, the strongest solutions provide document control with version management, metadata-driven organization, workflow automation, and audit trails, all integrated into a unified environment.
For life sciences, this means:
- Structured metadata from the start: Documents are tagged with regulatory attributes (module, section, content type, classification level) as they enter the system, enabling automated routing and search
- Audit trail automation: Every action – creation, review, approval, change, is automatically recorded to satisfy 21 CFR Part 11 and EU Annex 11 requirements without manual documentation
- Role-based access controls: Reviewers, approvers, and stakeholders see only content they’re authorized to view and can only perform actions within their scope
- Version control by design: The system tracks every iteration, preventing confusion about which version is current and approved
Principle 2: Automate Metadata Enrichment and Data Validation
One of the most time-consuming steps in traditional submissions is ensuring that all required data is present, properly formatted, and compliant with regulatory standards. McKinsey’s research on AI in regulatory submissions shows that early pilots using gen-AI-assisted approaches can reduce clinical study report authoring time by 40 percent while simultaneously improving accuracy.
Modern AI-enabled platforms accomplish this by:
- Auto-generating metadata: Machine learning models analyze document content and automatically assign regulatory classifications, data types, and references without human intervention
- Flagging completeness issues: Systems cross-check submissions against regulatory checklists and highlight missing data, incomplete sections, or formatting errors before human review even begins
- Validating data integrity: AI validates cross-references, checks for contradictions between modules, and ensures that all required fields are populated
- Reducing first-cycle deficiencies: By catching problems proactively, organizations minimize requests for additional information (RFIs) from health authorities
The cumulative effect: teams spend less time on data wrangling and more time on clinical interpretation and regulatory strategy.
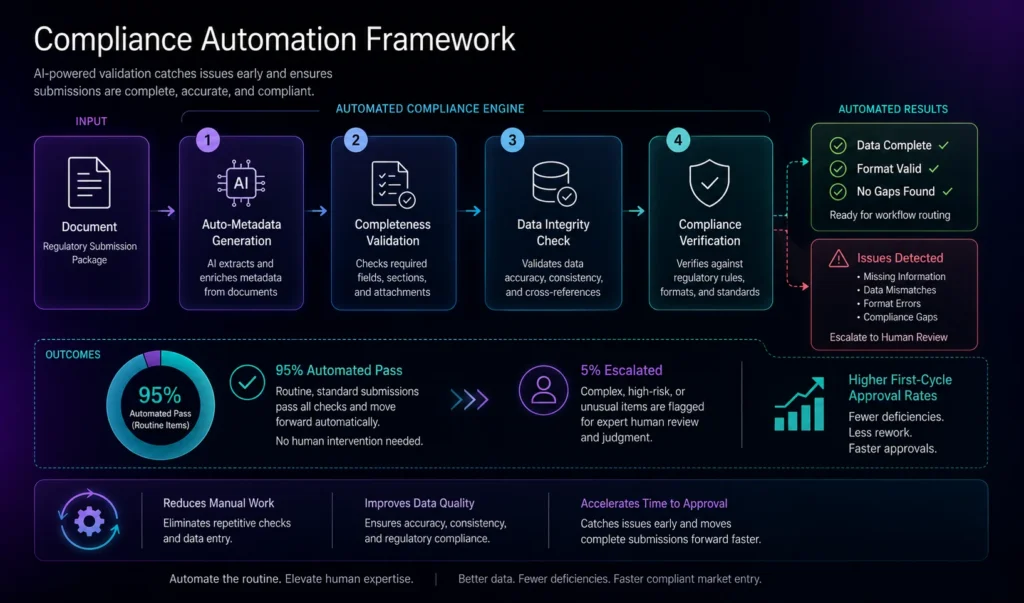
Principle 3: Embed Workflow Automation and Parallel Processing
Traditional approval processes move sequentially—one team completes review, passes to the next, and waits. Leading organizations restructure workflows to enable parallel review, reduce handoffs, and automate routine tasks.
Key workflow improvements include:
- Parallel review tracks: Different teams (regulatory, quality, medical, legal) review their respective sections simultaneously rather than sequentially, cutting total cycle time significantly
- Smart routing: Submissions automatically route to the appropriate reviewer based on document type, risk level, and expertise, eliminating delays from manual assignment
- Approval gating: Workflows trigger automatic holds if compliance checks fail, preventing incomplete submissions from advancing to the next stage
- Escalation automation: If a review exceeds a defined timeframe, the system automatically escalates to management, preventing bottlenecks from going unnoticed
According to McKinsey’s analysis of pharma submission excellence, leading companies have compressed submission timelines from standard 90-180 day cycles to as little as 12-16 weeks for their entire portfolios through such redesigns.
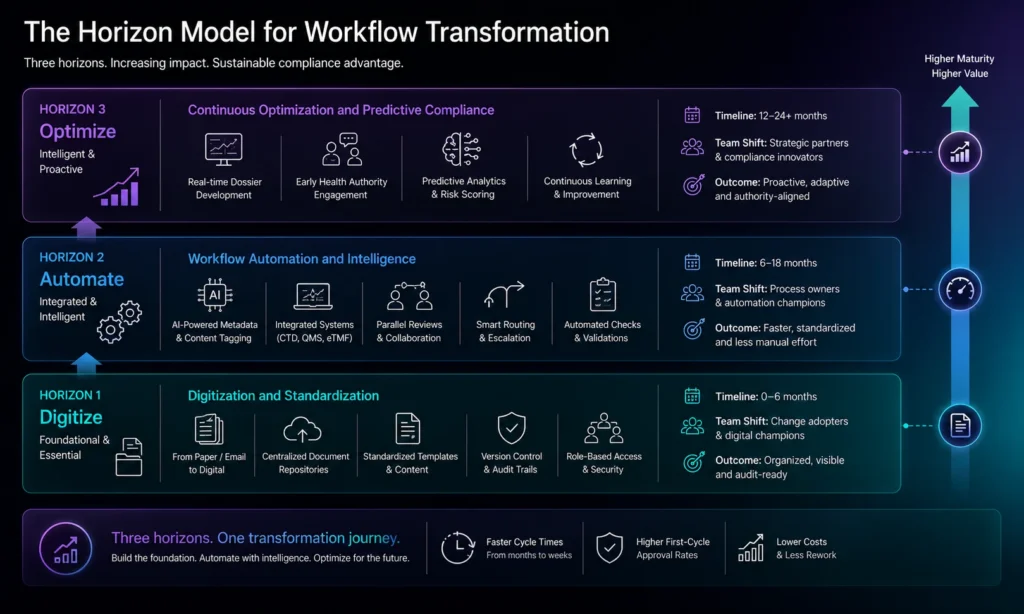
From Bottleneck to Compliance Engine: The Transformation Model
The shift from traditional approval processes to modern compliance engines involves three horizons of change:
Horizon 1: Digitize and Standardize Current Processes
The first step is moving from paper and email-based workflows to digital systems with standardized data structures. This means:
- Implementing centralized document repositories with version control and audit trails
- Establishing standard metadata schemas that all teams use consistently
- Creating digital templates for common submission types (IND, BLA, NDA, supplements)
- Training teams on new digital workflows and governance expectations
Horizon 2: Automate and Integrate Across Functions
Once processes are digital, automation becomes possible. This involves:
- Deploying AI to auto-generate and validate regulatory metadata
- Integrating submission systems with clinical data repositories, manufacturing systems, and quality management systems
- Automating compliance checks and completeness validation
- Enabling parallel review workflows with smart routing and escalation
Horizon 3: Optimize for Speed and First-Cycle Success
The most mature organizations treat regulatory submissions as a continuous process integrated with clinical development, not a final scramble before filing. This means:
- Building submissions incrementally as data becomes available (real-time dossier development)
- Engaging health authorities early using structured data and pre-submission interactions
- Using predictive analytics to identify potential approval risks and address them proactively
- Continuously learning from submission outcomes to improve future processes
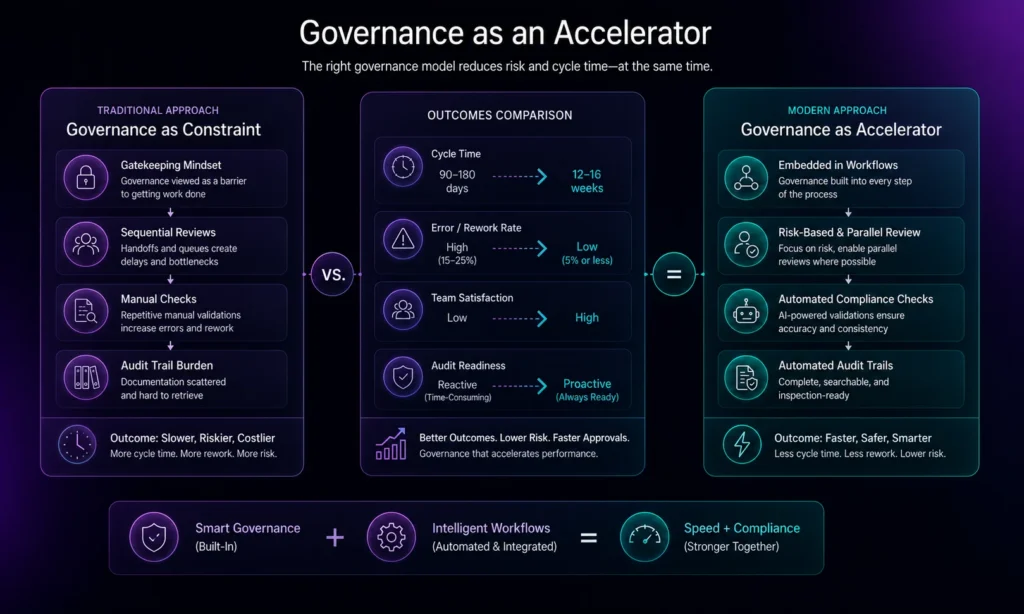
Governance as an Accelerator, Not a Constraint
A critical mindset shift separates organizations that successfully accelerate approvals from those that remain bottlenecked: viewing governance not as a hurdle to get through, but as a platform that enables speed and risk management simultaneously.
Traditional governance is often experienced as constraint with multiple reviews, layers of approval, gatekeeping. Modern compliance engines reverse this dynamic:
- Governance embedded in workflows: Approval rules are encoded into the process itself, not applied as separate review steps. Missing data triggers automatic holds before review even begins
- Risk-based review intensity: Not all content requires equal scrutiny. High-risk sections (claims, safety statements, manufacturing processes) receive rigorous review; routine sections move quickly through streamlined approval
- Clear visibility and accountability: Every team sees submission status in real time, knows which documents await review, and can identify bottlenecks immediately
- Audit trail as asset, not burden: Rather than scrambling to reconstruct who approved what and when, the system maintains automatic, audit-ready documentation of every decision
As Aprimo explains in its perspective on using AI safely for compliance and governance, successful organizations embed governance directly into content operations rather than treating it as separate oversight. This approach reduces manual compliance work while simultaneously strengthening audit readiness.
Implementation Roadmap: From Bottleneck to Engine
Organizations looking to transform their approval workflows should follow this structured approach:
Phase 1: Assess and Plan
- Document current submission processes and cycle times
- Identify the biggest sources of delay (missing data, rework, handoffs, bottleneck points)
- Define target improvements (e.g., reduce 60-day review cycle to 30 days)
- Engage stakeholders across regulatory, quality, legal, medical affairs, and clinical operations
Phase 2: Implement Centralized Platform
- Deploy a centralized document management system designed for regulated industries
- Configure metadata schemas, approval workflows, and compliance rules
- Set up role-based access controls and audit trail logging
- Train all users on new processes and system navigation
Phase 3: Automate Routine Processes
- Implement automated metadata tagging and validation
- Deploy AI-powered completeness checks and data validation
- Set up smart routing and escalation workflows
- Test parallel review processes with high-volume document types
Phase 4: Optimize and Integrate
- Integrate with clinical data systems and quality management systems
- Implement analytics to track cycle times and identify remaining bottlenecks
- Establish real-time dossier development processes
- Train teams on advanced features and optimization techniques
Results: What Leading Organizations Achieve
Organizations that successfully transform approval workflows report measurable improvements:
Time Compression: Cycle times reduce from 50-60 days (or longer) to 20-30 days or less. McKinsey’s documented case of submission timeline compression shows leading companies achieving 50-65 percent reductions.
Error Reduction: Automated compliance checks catch issues before human review. The Merck-McKinsey case study documented 50 percent reduction in CSR authoring errors.
First-Cycle Approval Rates: When submissions are complete, accurate, and well-organized, approval rates improve. Fewer requests for additional information (RFIs) mean fewer delays and faster patient access.
Regulatory Relationship Improvement: Health authorities notice organizations submitting high-quality, complete dossiers. This builds trust and can support innovative review pathways and expedited approvals.
Team Satisfaction: Removing manual, repetitive work improves job satisfaction for regulatory professionals, allowing them to focus on strategic aspects of regulatory strategy rather than administrative overhead.
Scalability: With efficient processes, organizations can manage larger portfolios without proportional increases in headcount. This proves critical as companies pursue multiple indications or global submissions.
Key Considerations for Life Sciences Organizations
When implementing compliance engines for approval workflows, life sciences organizations must account for unique industry requirements:
Regulatory Compliance Requirements: Solutions must support 21 CFR Part 11 (electronic records and signatures), EU Annex 11 (computerized systems), and the evolving technical standards of global health authorities (FDA’s eCTD, EMA’s CTD, etc.).
Cross-Functional Coordination: Approvals require alignment between regulatory affairs (data completeness), quality (manufacturing compliance), medical affairs (clinical interpretation), legal (intellectual property and risk), and clinical operations (study data integrity). Platforms must facilitate seamless collaboration across these functions.
Global Complexity: Life sciences organizations submit to multiple health authorities with different requirements, languages, and regulatory preferences. Solutions must support global templates, translations, and jurisdiction-specific workflows.
Audit Readiness: Regulatory submissions are auditable events. Systems must maintain comprehensive audit trails that satisfy FDA 483 observations, inspection findings, and internal quality audits without burdening teams with manual documentation.
Integration with Existing Systems: Organizations typically have invested in clinical trial management systems (CTMS), quality management systems (QMS), laboratory information systems (LIMS), and enterprise data warehouses. New approval platforms must integrate cleanly with these existing systems.
Conclusion
The transformation from regulatory bottleneck to compliance engine is no longer aspirational—it is achievable and increasingly urgent. As the cost of drug development climbs and patient populations wait for new therapies, every day saved in approval cycles translates to competitive advantage and, more importantly, to earlier patient access.
The organizations leading this transformation share common characteristics: they view governance as a speed enabler rather than a constraint; they embed compliance into workflows rather than applying it as a separate review layer; they leverage automation to reduce manual work while strengthening audit readiness; and they treat approval processes as integrated extensions of clinical development rather than isolated downstream activities.
The path forward is clear. Organizations that move quickly to centralize document management, automate compliance checks, and redesign workflows for parallel processing will compress approval cycles, reduce errors, and build stronger regulatory relationships. Those that remain dependent on manual processes, fragmented systems, and sequential reviews will face mounting pressure as competitors pull ahead.
The bottleneck is not inevitable. It is an opportunity—to redesign how life sciences teams collaborate, approve, and accelerate the delivery of life-saving therapies to patients who need them most.
FAQ
What is the typical current timeframe for regulatory submissions in pharma, and why is it so long?
Traditional submission timelines range from 90-180 days from clinical database lock to filing-ready dossier, and even longer for complex products. Delays stem from fragmented documents across multiple systems, incomplete submissions requiring rework, sequential handoffs between regulatory/quality/medical teams, and manual compliance verification. Leading organizations have compressed this to 12-16 weeks through centralized systems and workflow automation.
How much can AI actually reduce submission preparation time?
McKinsey’s collaboration with Merck demonstrated that AI-powered clinical study report (CSR) authoring reduced first-draft writing time from 180 hours to 80 hours while cutting errors by 50 percent. Additionally, gen-AI-assisted medical writing can reduce end-to-end CSR authoring cycle time by 40 percent. These gains come from automating routine writing, validating data completeness, and flagging inconsistencies before human review.
What regulatory compliance requirements must approval systems satisfy?
Life sciences systems must comply with 21 CFR Part 11 (electronic records and signatures), EU Annex 11 (computerized systems in regulated environments), and FDA/EMA regulatory guidance on electronic submissions. Systems must maintain complete, unalterable audit trails, enforce electronic signatures, support role-based access controls, and enable organizations to pass FDA inspections and demonstrate system validation.
How do I know if my organization is ready to implement a compliance engine system?
Readiness indicators include: documented approval cycle times (baseline metrics), cross-functional agreement that approval is a bottleneck, executive sponsorship for process redesign, IT infrastructure capable of supporting integrated systems, and available budget for platform investment and change management. Start with a process assessment to identify your biggest delays before selecting a solution.
What is the ROI on investing in centralized approval systems?
ROI drivers include: reduced cycle times (faster revenue recognition and patient access), fewer first-cycle deficiencies (avoiding costly RFI cycles), reduced error rework, improved team productivity (shifting from administrative work to strategy), and scalability (managing larger portfolios without proportional headcount increases). The Merck case showed dramatic productivity gains; organizations typically see positive ROI within 12-18 months as cycle time compression accumulates across multiple submissions.
